南湖新闻网讯(通讯员 欧阳傲天)近日,我校农业微生物资源发掘与利用全国重点实验室、动物育种与健康养殖前沿科学中心、湖北洪山实验室、动物医学院周红波教授团队在流感病毒与宿主相互作用机制研究领域取得重要进展。相关成果以“The Hemagglutinin of Influenza A Virus Induces Ferroptosis to Facilitate Viral Replication”为题在Advanced Science期刊上发表。
A型流感病毒(Influenza A virus, IAV)是一种有囊膜、分节段的单股负链RNA病毒,是重要的人兽共患病原体。流感病毒感染可引起急性呼吸道疾病,导致每年的季节性流行和偶发的全球大流行,对人类和动物健康构成重大威胁。IAV被证实可诱导多种类型的细胞死亡,包括细胞凋亡、坏死、细胞焦亡和自噬。在病毒感染过程中,细胞死亡一方面可以促进病毒致病性,另一方面保护宿主免受进一步感染。因此,IAV调控细胞死亡机制的解析,对于阐释病毒复制策略以及病毒防控具有重要意义。
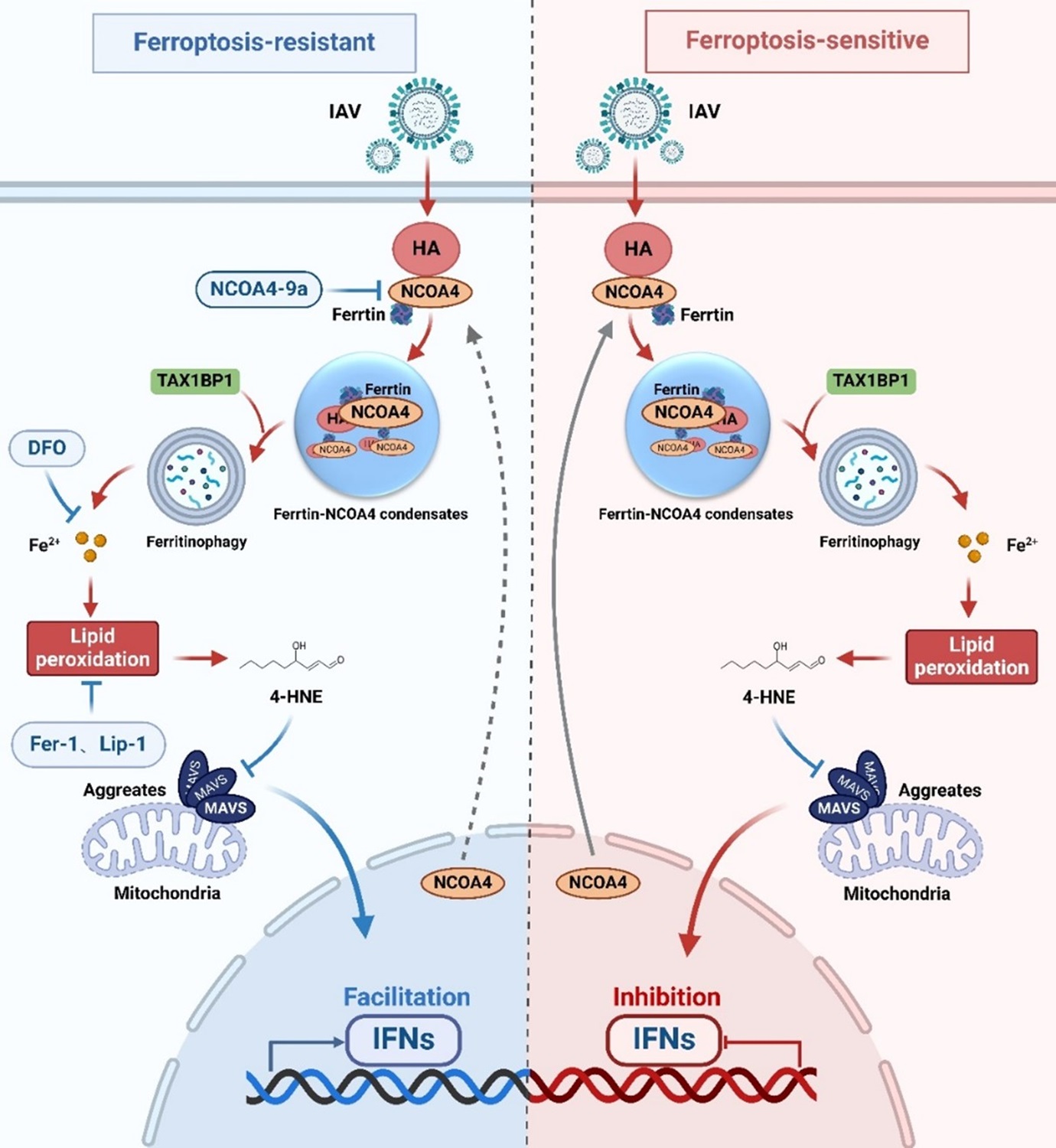
IAV血凝素通过铁自噬诱导脂质过氧化抑制MAVS介导的抗病毒免疫
此前团队研究发现,IAV可通过病毒蛋白M2、PB1-F2和NP诱导细胞自噬,进而调控病毒自身的复制和致病(Autophagy, 2019 & 2020;Journal of Virology, 2019)。那么其他细胞死亡形式是否也参与调控IAV复制呢?铁死亡是一种非凋亡细胞死亡形式,其本质是细胞内Fe2+的累积,引起芬顿(Fenton)反应,催化多不饱和脂肪酸磷脂(PUFA-PLs)发生脂质过氧化,导致脂质过氧化物累积,破坏细胞膜结构,最终导致细胞死亡。虽然有报道表明IAV可诱导细胞铁死亡,但IAV诱导铁死亡的分子机制,以及铁死亡对病毒复制的影响仍未深入阐明。
本研究围绕IAV诱导的铁死亡展开探索。研究首先确定了IAV诱导的呼吸道细胞铁死亡;同时,抑制铁死亡不仅抑制了IAV诱导的细胞死亡,也显著抑制了病毒复制。随后利用基因敲除等手段,发现IAV血凝素蛋白是诱导铁死亡的关键因素。通过质谱鉴定发现血凝素蛋白通过与NCOA4和TAX1BP1相互作用诱导铁自噬,促进铁蛋白-NCOA4聚集体的形成,进而诱导铁死亡。进一步研究发现,血凝素蛋白诱导铁自噬导致细胞脂质过氧化,脂质过氧化产物(如4-HNE等)增多,从而抑制MAVS的聚集,损害由MAVS介导的抗病毒免疫。最后,利用腺病毒表达系统在体内验证了血凝素蛋白在小鼠肺部也诱导了铁自噬并损害MAVS介导的抗病毒免疫。
本研究揭示了IAV血凝素蛋白诱导铁自噬导致细胞脂质过氧化,抑制MAVS介导的抗病毒免疫的新机制,为抗IAV策略的开发提供了潜在的靶标。
英文摘要
Ferroptosis is a novel form of cell death caused by the accumulation of lipid peroxides in an iron-dependent manner. However, the precise mechanism underlying the exploitation of ferroptosis by influenza A viruses (IAV) remains unclear. The results demonstrate that IAV promotes its own replication through ferritinophagy by sensitizing cells to ferroptosis, with hemagglutinin identified as a key trigger in this process. Hemagglutinin interacts with autophagic receptors nuclear receptor coactivator 4 (NCOA4) and tax1-binding protein 1 (TAX1BP1), facilitating the formation of ferritin-NCOA4 condensates and inducing ferritinophagy. Further investigation shows that hemagglutinin-induced ferritinophagy causes cellular lipid peroxidation, inhibits aggregation of mitochondrial antiviral signaling protein (MAVS), and suppresses the type I interferon response, thereby contributing to viral replication. Collectively, a novel mechanism by which IAV hemagglutinin induces ferritinophagy resulting in cellular lipid peroxidation, consequently impairing MAVS-mediated antiviral immunity, is revealed.
原文链接:https://onlinelibrary.wiley.com/doi/10.1002/advs.202404365
审核人:周红波
